
Step-by-step guide to FluoroSpot
Published: October 10, 2022
Updated: December 10, 2025
21 minute read
Authored by: Jens Gertow
FluoroSpot is the multiplex version of ELISpot. The assay combines the sensitivity of ELISpot with the capacity to study secretion of several analytes simultaneously. This is an in-depth guide for each step of the FluoroSpot protocol, including tips and tricks, key details to think about, and best practice.
FluoroSpot is an immunoassay used to quantify analyte-secreting cells. Just like in the ELISpot assay, target proteins secreted by cells are captured by specific antibodies immediately after secretion and throughout the stimulation process, making it an extremely sensitive sandwich assay. The difference to ELISpot lies in the mode of detection: where ELISpot relies on an enzymatic reaction, FluoroSpot utilizes fluorescence. This opens up for multiplex analyses of several analytes simultaneously, enabling studies of cell populations with different functional profiles.
We developed the technique in 2008 and have continually launched new kits and refined the protocol. Thus, we can offer many tips to fine-tune and maximize the success of your assay. We will discuss such suggestions in this guide, starting where the assay begins – with coating the FluoroSpot plate – and continue through the steps of the assay.
The general steps of a FluoroSpot assay


1. Coat the plate with capture antibody
Steps under section A in our FluoroSpot Flex protocols

A FluoroSpot plate is, just like an ELISpot plate, a plastic microplate with a membrane made of polyvinylidene difluoride (PVDF) in the bottom of each of the 96 wells. To avoid problems with autofluorescence, the PVDF membrane of the FluoroSpot plate has been made low-fluorescent. Apart from the membrane, this plate, called IPFL, has the same configuration and characteristics as the clear MSIP plate used for ELISpot. The IPFL plate is included in all our FluoroSpot kits, but we can unfortunately not sell it separately.
A key feature of the IPFL (MSIP) plate is that it has one “cut” corner top left.


Ethanol pre-treatment and plate coating – be meticulous
The PVDF membrane can only bind the required amount of capture antibody after ethanol pre-treatment which makes the membrane hydrophilic. In addition, the spot appearance is optimized by using the recommended concentration of capture antibody (this might differ depending on analyte, but is usually 15 µg/ml). If you dilute the capture antibody to lower than recommended concentrations, you risk getting fuzzy spots that are hard to count.


Ethanol pre-treatment is pivotal if you are coating the IPFL plate yourself
Higher concentration of capture antibody à More efficient capture of analyte à More distinct appearance of spots à Improved spot count


Diluting the capture antibody leads to fuzzy spots
How to coat a FluoroSpot plate
1. Add 20 µl freshly prepared ethanol (diluted to 35% from 99.5% stock in sterile water) to all wells you plan to coat. Upon contact with ethanol, the membrane will instantly turn pale grey, indicating that the PVDF has been activated. Leave the ethanol in the wells for no longer than 1 minute. If you intend to coat several plates, treat one or two plates at a time to stay within the short incubation time for ethanol.
2. Add sterile water to the wells without removing the ethanol, then wash 5 times with sterile water (200 µl per well).
3. Empty the plate. Add coating antibody diluted in phosphate-buffered saline (PBS; 100 µl per well). We generally recommend a concentration of 15 µg/ml coating antibody (1.5 µg antibody per well), but check the product-specific protocol for details. Leave the plate with coating antibody solution covered at 4-8 °C overnight.
4. The following day, decant the coating solution and wash 5 times with sterile PBS. Use the plate immediately for the FluoroSpot assay.
NB! In FluoroSpot, it is common to coat with a mixture of capture antibodies with different specificities, e.g., mAbs to IFN-γ, IL-2, and granzyme B. Regardless of number of coating antibody clones, the total volume should be 100 µl per well. Thus, for three coating antibody clones, dilute all to e.g., 15 µg/ml in the same tube, and add 100 µl of that mix to each well.
Tip! The ethanol pre-treatment and plate coating steps are omitted if you choose pre-coated FluoroSpot plates.


The total volume of the mix should be 100 µl
Handle the plate carefully
The following tips are applicable for all steps of the assay:
-
Do not allow the plate membrane to dry out during the assay. If your plate membrane does dry out during ethanol pre-treatment – don’t panic, simply repeat the ethanol pre-treatment steps.
-
Handle plates gently and be careful not to put pressure on the underside of the plate. Grip the plate by the edges, not the top or bottom, and avoid excessive force when emptying it.
-
Do not allow the pipette tip to touch the membrane, as it may damage the membrane and lead to artifacts.
-
Ensure that the dispensed solutions/reagents cover the wells. Also avoid bubble formation when pipetting, as bubbles may hinder reagents from reaching the bottom of the well. Incomplete solution cover or bubble formation may result in blank wells after development.
How to handle the underdrain of FluoroSpot (MSIP) plates
-
Wash plates using a multichannel pipette when you need to work in a sterile environment, e.g., in a laminar flow hood. When sterile conditions are not required (please check the protocol of the product), you can use an ELISA plate washer but make sure to adapt the washer head position to the shallower wells of FluoroSpot plates.
-
Do not use Tween 20 in your wash buffer. We know it’s commonly included when running ELISA assays, but please don’t include it for FluoroSpot. Tween 20 may damage the PVDF membrane and/or solubilize cell membranes, both of which will negatively affect spot formation and appearance.
-
If you need to dissolve your antigen of interest in DMSO, make sure that the DMSO in the cell culture is diluted to a working concentration of below 0.5%. DMSO at a higher concentration may damage the PVDF membrane as well as the cells, and cause artifacts.
-
Avoid splashes when emptying plates, as liquid might splash back into the wells. As a final emptying step, tap the plate gently against a paper towel.
2. Add cells and stimuli to the plate
Steps under section B in our FluoroSpot Flex protocols

Whether you coat the plates yourself or run pre-coated plates from us, by this stage the FluoroSpot plate is almost ready for your cells. One first needs to condition the plate by adding 200 µl of the same medium as your cells for 30 minutes at room temperature. During this time you can start preparing your cells and stimuli. Being a cell-based assay, cell-viability is key to a successful FluoroSpot assay.
Cells are incubated in the presence or absence of an activating stimulus (an antigen or polyclonal stimulatory agent) at an appropriate temperature (usually 37 °C). The incubation period should allow for optimal analyte secretion and thus can last from a few hours up to a few days. We recommend an assay setup with triplicate wells for all conditions.
Most cell sources can be used – as long as the cells are alive
FluoroSpot can be applied to basically any cells to investigate protein secretion. We have seen publications with a wide range of cells, including human blood cells, mouse spleen cells, cell cultures, cells from mucosal tissue, heterogeneous cell populations and purified homogeneous cells, and cell clones. The most commonly used cells, at least from human samples, are peripheral blood mononuclear cells (PBMCs). They can be obtained from blood by density centrifugation using e.g., Ficoll. The resulting PBMCs can be used directly or may be frozen for later use.
Freezing:
Standard freezing procedures include the use of DMSO and FCS in the medium. We first put the vials with cells in a freezing container at -80 °C overnight and then transfer to liquid nitrogen or a freezer at -150 °C.
Thawing:
After washing the cells twice (to get rid of the DMSO), the cells should be rested in fresh medium for 1 hour at 37 °C. This allows for removal of cell debris, which will improve assay performance.
Cell quality is crucial for reliable results
Using samples with a high proportion of viable cells and a low proportion of apoptotic or dead cells will give you the greatest likelihood of success. This is not unique for FluoroSpot, but true for any cell-based assay. If your sample contains a high proportion of dead cells, you cannot rely on your final results as they are based on the ratio of responsive cells (spot-forming units) to total cells.
Freezing and thawing cells is not a problem if you do it right. But an important note is that PBMCs should be prepared as soon as possible after blood collection. This is because cells prepared from blood samples older than around 8 hours may be contaminated with activated granulocytes which can interfere with the results. If blood samples must be stored before preparation (for example over-night if you received the sample late in the afternoon), high-quality PBMCs can be obtained by:
- Gently agitating the blood samples
- Diluting the blood samples 1:1 in PBS or RPMI
- Depleting the granulocytes from the blood samples using commercial depletion kits, e.g., from Stemcell Technologies
Choose a cell culture medium appropriate for your cells
The cell culture medium plays a crucial role in the well-being of your cells. We recommend RPMI 1640 with L-glutamine and 10% fetal calf serum (FCS) for most cells. At Mabtech, we usually supplement the medium with HEPES to maintain the pH of the cell culture and antibiotics to decrease the risk of contamination. Since RPMI 1640 might not be the optimal medium for your experiment, you must do the homework of checking which medium is best for the cells you are studying.
Generally, we recommend FCS as the source of serum. Different serum batches may vary in their content and functionality, and it’s wise to test a new serum batch before use in FluoroSpot. The serum used should be selected to support cell culture and give low background staining, i.e., should not itself give rise to cell activation or protein secretion. We advise you not to use homologous serum (e.g., human serum for human samples), as it might contain the analyte you intend to detect and/or heterophilic antibodies, both of which may cause background noise.
Our in-house cell culture medium:
-
RPMI 1640
-
10% FCS
-
2 mM L-glutamine
-
10 mM HEPES
-
100 µg/mL penicillin
-
100 µg/mL streptomycin
If FCS cannot be used in your assay, it is possible to use a serum-free medium containing defined proteins as a substitute for serum. Of all serum-free media we have tested, AIM-V works best for FluoroSpot.
Regardless of which cell culture medium you choose, it’s essential to use the same medium throughout the assay, including the blocking/conditioning step before adding the cells. Using the same media ensures the cells' environment remains constant in each step, reducing the number of variables that could affect the final results through non-specific stimulation.
Choose a suitable cell number
At this point, the plate has been coated with capture antibody and conditioned with cell culture medium for at least 30 minutes at room temperature. Before adding cells to the plate, you need to count and adjust them to the required concentration in cell culture medium.
Cell counting can be done with an automated cell counter or trypan blue staining using a microscope. Because only living cells will be able to secrete the analyte, make sure to exclude dead and, if possible, apoptotic cells from the cell count. If the cell concentration turns out to be lower than required, you will need to concentrate the cells by centrifugation.
The following should be considered:
-
If unsure, start with 250,000 cells per well
The number of cells in each well must be adapted to the cell type and the expected frequency of secreting cells: The lower the expected frequency of responding cells, the higher the number of cells per well. If the expected frequency is 1 in 10,000 cells, you will need approximately 250,000 cells per well. Conversely, when the frequency is high, for example 1 in 100 cells, it’s sufficient to seed 50,000 cells per well. In general, antigen-specific responses require higher cell numbers (e.g., 250,0000 cells per well) than polyclonal/mitogenic positive controls (e.g., PHA, 50,000 cells per well). -
Cells “like” to be in contact with other cells
The number of spot-forming units measured in FluoroSpot does not always correlate with the total number of cells because a minimum number of cells is required for proper cell-to-cell contact and thus optimal stimulation. For example, when analyzing antigen-specific T cells in a PBMC sample, total cell numbers should not be less than 200,000 cells per well. -
Avoid vortexing
Resuspend cells and pellets gently using a pipette. -
It is sometimes beneficial to stimulate cells before adding them to the FluoroSpot plate
For analytes that need a longer time for secretion to be initiated, it is possible to pre-stimulate the cells (preferably in round-bottom polypropylene tubes or plates to augment cell contact) before adding them to the FluoroSpot plate. This is good practice in certain circumstances, e.g., when stimulating memory B cells for 72 hours with R848 + IL-2 to facilitate differentiation into immunoglobulin-secreting plasma cells. If you choose pre-stimulation, it is important to note that cells must be washed before addition to the FluoroSpot plate to avoid background staining of the membrane.
Seed and stimulate the cells
When you have chosen the cell number that is appropriate for the experiment, it’s finally time to seed the cells onto the plate and stimulate them. In addition to your stimulus of interest, we recommend you to include three control conditions:
Positive controls
Antigen-specific or polyclonal
-
Indicate if the assay works and the cells are functional
-
Reveal false negative results
Negative controls
Cells without stimuli
-
Indicate number of spontaneously secreting cells
-
Reveal false positive results
Background
No cells, but all other reagents
-
Indicate if reagents induce false spots (aggregates)
-
Reveal false positive results
Choosing the relevant compounds for control stimulation is largely dependent upon the cell type and the nature of your experiment. Stimuli can be monoclonal antibodies (anti-CD3 for T cells) or peptides (CEFRAS Global peptide pools for CD8+ and CD4+ T cells), synthetic compounds (R848+IL-2 for B cells) or substances purified from bacteria or plants (PHA for T cells). Antigen-specific stimulation using peptide pools or monoclonal antibodies are preferable in our view, since these reagents are well defined. More on peptide pools and polyclonal stimuli here.
For all types of stimuli, antigen-specific or polyclonal, we recommend either of the following two techniques:
I. Add stimuli first, then cells
With this approach, stimuli and cells are added separately to the plate. When equal volumes of stimuli and cells (50 µl each) are added sequentially to each well, the stimuli and cell suspensions should be prepared at 2x the final assay concentration. A cell concentration of 5x106 cells/ml will give you 250,000 cells per well. It is crucial to add the stimuli first and then the cells. Do not add anything to the wells after the cells have been seeded, as this could push the cells to the edges of the wells, leading to suboptimal results.
II. Mix the cells and stimuli in tubes before addition to the plate
Mix the cells and stimuli in tubes to the final assay concentrations. Add 100 µl per well; a cell concentration of 2.5x106 cells/ml will give you 250,000 cells per well. Because cells will continuously sediment in the tubes, remember to carefully resuspend the cell suspension throughout this procedure.


If your samples take up only a part of the plate, fill the surrounding wells with medium only (100 µl per well) to minimize evaporation from your samples during incubation.
Put the plate in the incubator
With the cells and stimuli added to the plate, the next step is to put the plate into an incubator. In general, the temperature of the incubator should be 37 °C with a CO2 level of 5%. Two things to think about when incubating cells:
-
Wrap the plate in aluminum foil
Most incubators maintain an optimal level of humidity, but it is good practice to wrap the plate in aluminum foil during incubation to reduce the risk of evaporation of the cell culture medium. -
Place the plates next to – not on top of – each other
Do not stack plates in the incubator. Stacking plates can lead to uneven conditions between the plates (e.g., temperature, CO2 level, evaporation rate), which may affect the cells and thereby the spot numbers.
3. Let the antibodies capture the analyte
Steps under section B in our FluoroSpot Flex protocols

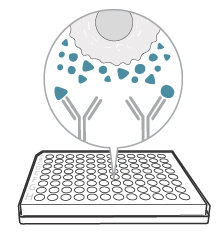
FluoroSpot uses specific capture antibodies in the bottom of the well to capture cytokines, immunoglobulins, or other target proteins immediately after secretion and throughout the stimulation process. This is what makes FluoroSpot so sensitive.
With the plate in the incubator, you can now just wait for the antibodies to capture the analytes. It is important to note that:
-
Different analytes have different kinetics
Consider the kinetics of secretion of the protein when establishing the optimal cell incubation time. Some analytes, such as granzyme B, are fully secreted only after more than 48 hours of stimulation, whereas others, such as IFN-γ, can be analyzed after only 12 hours. If you are studying both analytes, choose the longer incubation time. It will not have any negative effect on the “quicker” analyte. -
The plate must not be moved during incubation
Avoid moving the plates during the incubation, as this can negatively affect spot formation. Even small movements should be avoided, so close the incubator door gently.
Capture effects and co-stimulation
When capture antibodies with different specificities are coated together, capture of one cytokine may affect the secretion of other cytokines, a phenomenon called “capture effects” or “cytokine absorption effects”. For example, presence of IL-2 capture antibodies may result in reduced activation of T cells, as capturing of IL-2 decreases the amount of IL-2 available for the cells. Thus, downstream IL-2-dependent cytokine secretion may be dampened. Luckily, capture effects can often be counteracted by addition of an anti-CD28 mAb, providing a co-stimulatory signal to T cells and thereby restoring cytokine secretion (see example in figure below).
Depending on cells and stimuli used, optimization of anti-CD28 concentration may be necessary, since too high concentration can lead to elevated non-specific cytokine secretion. Also note that capture effects are more pronounced in positive controls than in antigen-specific responses.


Capture effect (left image) 1) IL-2 secreted by the activated T cell is captured by coated anti-IL-2 capture antibodies. 2) As a result, IL-2-stimulation of the T cell itself (autocrine stimulation) as well as nearby T cells (paracrine stimulation) is impaired, ultimately leading to (3) decreased granzyme B secretion.
Counteraction (right image) 1) An anti-CD28 antibody can be added to provide a co-stimulatory signal that can (2) restore for example granzyme B responses.
Our FluoroSpot Plus kits are evaluated for capture effects, and in most studies of T cell responses we recommend co-stimulation with anti-CD28. With FluoroSpot Flex, however, it is possible to combine analytes as you wish to build your own kit. Most analyte combinations work just fine, but there are a few combinations that we advise you not to use. For guidance look at our analyte combination table.
If your specific analyte combination isn’t in the analyte combination table, and you are unsure, you can investigate capture effects like this:
-
Coat wells with single capture antibodies, one set of wells per capture antibody.
-
Coat wells with a mixture of the capture antibodies.
-
Compare the number of spot-forming units in the different single conditions to the mixture condition. If you see a difference between the single coated wells and the mixture wells, you might have identified a capture effect.
-
Assess the compensatory effect of anti-CD28 mAb by comparing cells cultured with and without the anti-CD28 mAb in the same setup as steps 1-3 above.
Capture effects and its countermeasures can be perceived as a bit artificial, but it’s no different than e.g., blocking the Golgi apparatus when doing intracellular cytokine staining in flow cytometry. All in vitro assays are models of reality. It’s just about finding a setup that suits your scientific question, allowing you to detect differences between sample and control.
4. Add detection antibody
Steps under section C in our FluoroSpot Flex protocols

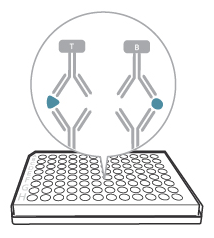
After the incubation step, the cells have secreted analytes in response to the stimulus, and the analytes have been captured by the specific antibodies coated on the plate. Time to detect. It might be obvious, but we’re saying it anyway: With FluoroSpot, you are detecting the footprint of the cell that was there, secreting the analyte you are studying. Thus, begin by washing away the cells: empty the plate and wash 5 times with PBS (200 µl per well).
FluoroSpot is based on a fluorescent detection system. Most analytes (except for immunoglobulins) are detected in a two-step fashion, where the first detection step is performed using a combination of a biotinylated and tag-labeled detection antibodies.
The tags and anti-tag antibodies we use are proprietary to Mabtech, and have been developed to work optimally in FluoroSpot.


Left: Two-step detection; Right: One-step detection
5. Add secondary detection reagent
Steps under section C in our FluoroSpot Flex protocols

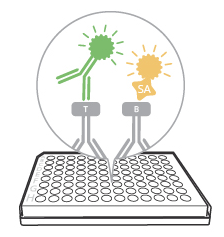
Once the detection antibodies have bound to the analyte, it’s time to add the secondary detection reagents, i.e., streptavidin and/or anti-tag antibody conjugated to fluorophores.
To increase the intensity of the fluorescent spots, our kits include a FluoroSpot enhancer solution. It’s a simple ready-to-use reagent used as a final step before analyzing the plate, easiest done like this:
How to use the FluoroSpot enhancer
-
Pour some FluoroSpot enhancer in e.g, a petri dish/reagent reservoir to allow the use of a multipipette.
-
Empty the last PBS wash by flicking the plate into a waste container.
-
Using the multipipette, add FluoroSpot enhancer (50 ul per well) and leave the plate at room temperature for 5-15 minutes.
-
Remove excess FluoroSpot enhancer by flicking the plate into a waste container and blotting against low-lint paper towels.
Important: Try to remove as much excess FluoroSpot enhancer as possible. In the video above, we tap the plate against paper towels. Since that recording, we have discovered that some brands of paper towels give off dust particles which can cause fluorescent artifacts. Thus, make sure to use low-lint paper towels and consider skipping altogether when using the 380 (blue, DAPI) detection system, as it is more sensitive to such particles.
6. Analyze the plate
Steps under section C in our FluoroSpot Flex protocols

You’re done with the wet-lab part, now it’s time to count the spots.
Plates need to be completely dry before analysis. If you are not in a hurry, leave the plate to dry overnight. To reduce the drying time to around 30 minutes, detach the underdrain carefully (using e.g., a pair of pliers) before placing them upside down in a laminar flow hood.
Fluorescent spots may fade due to excessive exposure to light, so keep the plates dark, and analyze them within one week of development.


FluoroSpot analysis requires an automated spot reader, like Mabtech IRIS™. The reader must be equipped with filters for the fluorophores used and selective for the specific wavelengths to avoid bleed-through artifacts. In addition, to obtain accurate measurements of cells secreting multiple analytes, the reader must identify spot centers and create an overlay analysis.
Mabtech IRIS utilizes RAWspot™ technology for accurate identification of spot centers and spot numbers. In addition, RAWspot provides information on relative spot volume.
What is a positive FluoroSpot result? Use the statistical method DFR for proper analysis.
When analyzing plates it is important to consider that, although many processes are automated in a reader, you are in control over which spots to count. With Mabtech Apex™, the software for ASTOR and IRIS, the default count settings are adequate for most situations. But, similar to gating populations in flow cytometry, you can use sliders for size and intensity to determine which spots you consider relevant.
In flow cytometry, one uses fluorescence-minus-one controls to get an idea of where to set the gate, whereas in FluoroSpot you decide on a count setting based on your experience. As FluoroSpot is so sensitive, it’s not uncommon to have spots in your negative control wells. That’s ok! We highly recommend to not subtract these spots from the “real” spots, but rather to use a statistical analysis where negative control spots are taken into account for when deciding on whether you are looking at a positive response. Please read more about this statistical method, called DFR, here.


Finally, when you have decided on a count setting, it’s good practice to choose a consistent count setting throughout the project (for all wells and plates treated in the same way).
For more information on IRIS and how to analyze plates in it, watch our video:
How to analyze FluoroSpot on Mabtech IRIS